Before beginning any project involving human subject’s research, all individuals on the research team (faculty, staff, and students) must complete a training certification program. UVU is contracted with CITI (Collaborative IRB Training Initiative) for customized training in the protection of human subjects in research. Your training will automatically be updated into the UVU IRB system after you have completed the program.
UVU has collaborated with CITI to make the IRB training much easier and more streamlined. If you have a UVU account, please use the following links to take you DIRECTLY to the trainings required for you to complete based on your role in the research.
Student Researchers click here
Faculty/Staff Researchers click here
There are seven required modules that all faculty/staff researchers will need to complete to earn this SBR IRB training certification. These seven modules should take approximately 2 to 3 hours to complete:
Researchers who do research with certain populations must also complete the applicable modules before submitting any related IRB protocols/packets. Faculty or staff members who work with students on research projects may also want to complete the Students in Research module (see Optional Modules) so they are familiar with the module their students will be required to complete. Optional modules include:
TO BEGIN: access the Faculty/Staff CITI training program.
All students who will be doing human subjects research at UVU are required to complete the "Student Researcher" certification on the CITI website. It should take you approximately one hour to complete. This will provide you with important information for you to become a professional and ethical researcher. There are two required modules:
TO BEGIN: access the Student Researchers CITI training program.
Campus Administrators are often researchers, or are in position where they must approve IRB packets submitted by their own faculty and staff. To ethically and thoroughly review these IRB submissions, administrators must understand ethical issues and challenges of human subjects' research and the applicable federal regulations. There are seven required modules that administrators will need to complete to earn this SBR IRB training certification. These seven modules should take approximately 2 to 3 hours to complete:
TO BEGIN: access the Campus Administrators CITI training program.
Faculty and staff serving on the UVU IRB must receive additional training to gain the depth and breadth needed to competently and professionally serve on the UVU IRB and review IRB submissions. In addition to the seven modules required of faculty/staff, IRB members must complete all 13 of the primary modules. IRB members may be reviewing IRB packets that deal with special populations or circumstances, it is imperative that they are knowledgeable about the IRB requirements of doing so. The complete training should take between 4-6 hours. The 13 modules include the following:
TO BEGIN: access the IRB Members and Staff CITI training program.
Planning and preparation are the key to a successful IRB application. If you have never submitted an application to the Utah Valley University Institutional Review Board before, this page will outline how to begin planning for your human subject’s research. It is the PI’s responsibility to oversee and ensure proper completion of information to the IRB including the original submission and all revisions requested during IRB review.
All researchers will need to have a clear project design before they are ready to submit their application to the IRB. A complete research plan includes:
A key role of the IRB is to assess the risks of the research to the participants. In order to approve research the IRB must find that:
The PI is responsible for identifying and describing risk to participants in their IRB application. They will also have to inform the participants of these risks including reasonably foreseeable discomforts, inconveniences, or harms in the informed consent documents.
When assessing the risks to participation in a study, the investigator should consider:
The PI should describe the potential benefits(s) that may be gained through this research. These benefits should be supported with sufficient evidence for the IRB to reach the same conclusion. The IRB will use this description to determine whether the risks of the research are reasonable in relation to the anticipated benefits. There are two main categories of benefits that may occur during a research study.
It is important to note that being compensated for research (e.g. money, gift card, extra credit in a course) is NOT considered a benefit for participating in the study.
Complete the Protocol Description Form found here. This form is required to be uploaded to your online application,
In order to assess the risks for study participants the IRB must review all material that participants will be exposed to. After the research plan is designed, applicants should generate all study materials required for recruitment and completion of the study itself. These include items such as:
If you are completing your research at an outside institution not affiliated with Utah Valley University please obtain documentation from them stating that they give permission for you to complete a research study at their location. This documentation must be included as part of a complete application for IRB review.
Informed consent is a basic ethical obligation and legal requirement for researchers working with human subjects. It is required to ensure study participants understand the research before they volunteer. This includes knowing what they will be asked to do as part of the study and appreciating the risks involved in participation.
An informed consent document is required to provide study participants with the information they need to decide whether or not to volunteer for a research study. The Utah Valley University IRB is moving away from offering consent document templates in order to allow for more flexibility in providing participants directed information related to each study. Consent documents must contain all the basic elements outlined in the Department of Health & Human Services (HHS) regulations (45 CFR part 46), and for some studies HHS regulations require additional elements to be included.
Basic Elements of Informed Consent
The UVU IRB provides a checklist of requirements to assist investigators in preparing their consent documents. You may download that checklist here.
The University of Utah’s IRB provides an excellent resource for researchers interested in learning more about each of the consent requirements here.
The consent document should clearly describe the research as it is presented in the IRB application. When writing your informed consent you should:
Under the HHS Common Rule, the IRB may approve an informed consent process that:
A waiver of documentation of informed consent means that the participants are provided with the required consent information, but the PI is not required to obtain the participant’s signature on the consent document. In order to approve a waiver of documentation of informed consent the IRB must determine that:
Waivers or alterations of informed consent elements can be requested for projects that are no more than minimal risk and involve the secondary analysis of data or projects involving deception. In order to approve a waiver or alteration of elements of consent the IRB must determine that:
You must indicate in your Axiom Mentor application if you are requesting a waiver of documentation or a waiver/alteration of consent elements. You will be required to provide additional information justifying the request.
Permission from one or more parents (for minors) or the legally authorized representative (for decisionally impaired individualsⱡ) is required through the informed consent process when those not able to provide consent are to be involved with research. However, while children/decisionally impaired individuals are not legally able to provide informed consent, they may possess the ability to assent or dissent from participation. Children/individuals should be asked if they are willing to participate in a research study if they are capable of understanding what is being asked of them. To determine whether a child/individual is capable of assenting the PI (and the IRB) should take into account the ages, maturity, and psychological state of the participants. Assent documents should:
ⱡPlease note that as a general rule, all adults, regardless of diagnosis or condition, should be presumed competent to consent unless there is evidence of serious mental disability that would impair reasoning or judgement. Assent is only required from persons formally adjudged to be decisionally impaired to consent. Federal guidelines do not provide standardized methods for determining capacity to consent. If working with subjects with cognitive or psychiatric disorders, the investigator should have a plan for assessing subjects’ ability to understand the nature of the research and information relevant to their participation; and to express a reasoned choice to volunteer or decline. The capacity to understand these concepts allows for an adult to consent to their participation without need for a legally authorized representative.
Please contact the IBR Office at [email protected] or schedule an appointment with the IRB Administrator (Appointments - sence (uvu.edu)) if you have questions about the consent process.
HOW TO USE THE NEW “CAYUSE” SYSTEM
The UVU IRB now uses a new third party system, Cayuse, for submission, review, and approval of IRB applications. Please click this Cayuse link to submit and renew all IRB proposals. Follow the steps below to submit your application for review:
STARTING AN APPLICATION:
[To see a short clip of how to begin a protocol submission in Cayuse, click here]
FILLING OUT THE APPLICATION SECTIONS:
PROJECT PERSONNEL
BASIC INFORMATION
STUDY DESIGN
STUDY SELECTION
STUDY PROCEDURES
SUBMITTING YOUR APPLICATION:
NEXT STEPS & REVISION REQUESTS:
After submitting your application, you will be brought to a new page. The bar at the top of the page shows the progression of your application. After certifying your application, that status should say “under pre-review.” Pre-review is the stage where your protocol will be reviewed by the IRB coordinator before being sent for final review by board member(s).
You will likely need to revise your protocol at certain points during the approval process – this is typical of the majority of protocols. You may receive revision requests during the pre-review phase by the IRB coordinator, and again during the review phase from board members. You will receive an automated email to notify you when you need to make revisions. The email will state that the submission has been “returned to investigators,” which means that the application has been sent back to you for changes and/or additional information. Instructions for how to address and respond to revision requests are below.
RESPONDING TO REVISION REQUESTS
You will receive an email when the person reviewing your submission has requested revisions from you.
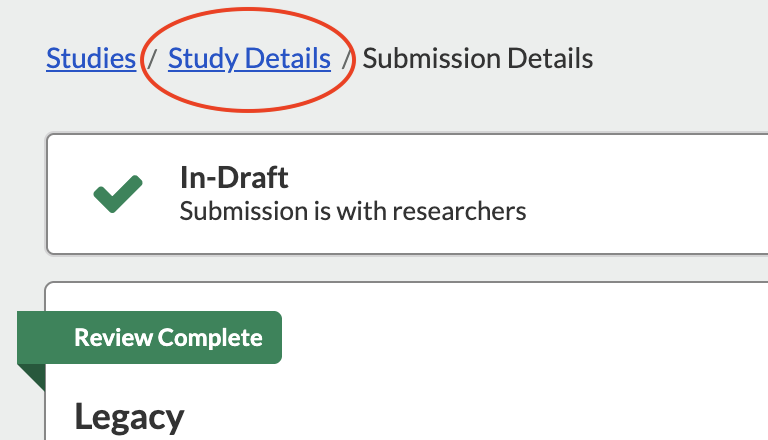
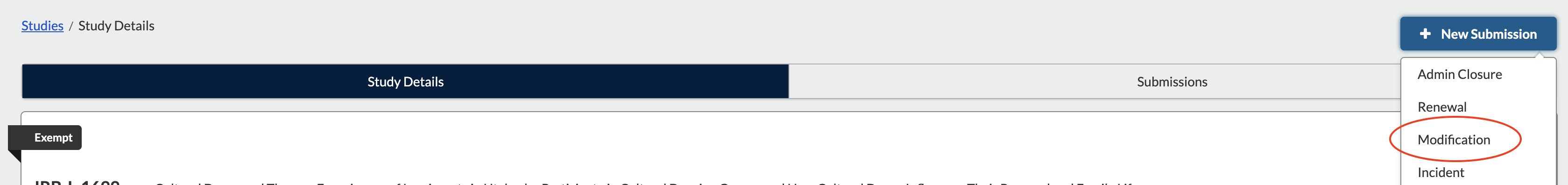
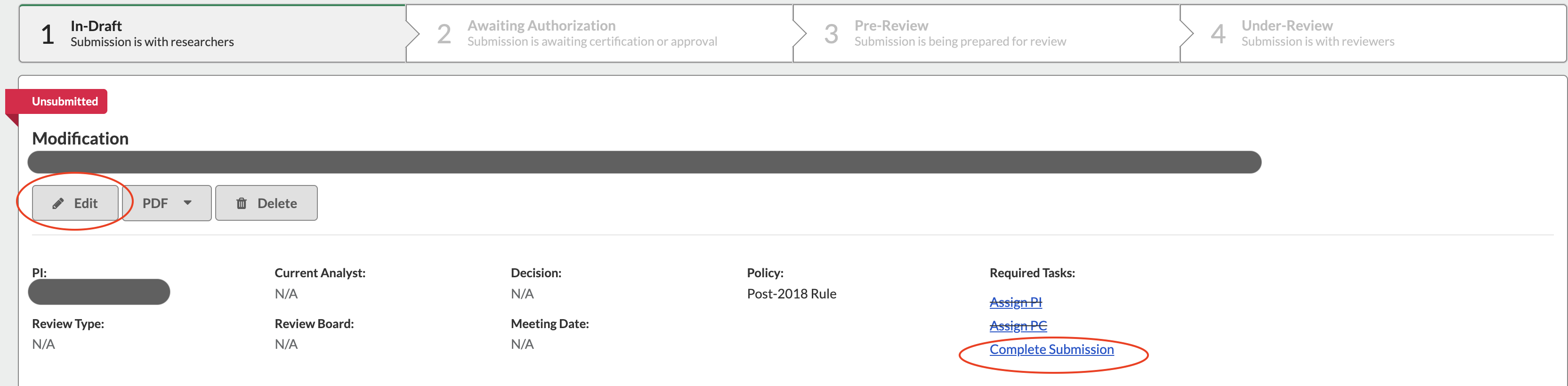
SUBMITTING AN AMENDMENT/MODIFICATION TO A LEGACY SUBMISSION
If you have worked with the IRB office to access a study you submitted in the old system, it will have been uploaded for you by an IRB staff member. The following steps tell you how to submit an amendment to your old protocol.